
NEXT GENERATION SYNTHESIS
Protocol
1. Resuspend oligos to make a 100 uM stock and then 10 uM dilutions that combine oligo pairs
-
Read nmol amount of oligo from tube label
-
For every nmol, add 10 ul water
-
Vortex or flick tube and wait several minutes for the oligo to go into solution
2. In a separate tube combine 80 ul water and 10 ul of each 100 uM oligo in a pair to make 10 uM paired dilutions
-
Oligo pairs that will be annealed as duplexes are named with the format X_s1 and X_s2. As an example, for X=T500:
-
Oligo pairs that will be used together as PCR primers together are named with the format XGolgat_1F and X_Golgat##R or XY_Golgat_1F and X_Y'_Golgat##R.
3. Prepare Golden Gate annealed duplex fragments.
-
In separate PCR tubes, mix the reagents
-
Incubate mix at 37C for 1 hr
-
Heat mixtures at 95C in thermocycler for 5 min and gradually cool to RT for 1 hr
4. Prepare the other Golden Gate fragments by PCR from plasmid X in primer pair X_Y_Golgat.
-
In separate PCR tubes, mix the 50 ul PCR reactions
-
Run the PCR program in your thermocycler with the lid set to 105 C
-
Run finished PCR reaction on a 1% agarose TAE gel (see protocol homework 2)
-
Purify the correct-sized band with a gel extraction kit (see kit manufacturer’s protocol)
5. Combine and assemble the Golden Gate fragments
-
For each fragment from step 4 derived from pGreen_c(G/C/B)FP_GolGat_1F, mix the following in a thermocycler tube
-
Run the assembly program in your thermocycler with the lid set to 105 C
6. Heat shock Golden Gate assembly into chemically competent E coli cells
-
Thaw pre-aliquoted 25 ul E coli cells on ice for 30 min
-
Add 2 ul of assembly to cells and keep on ice for 15 min
-
Heat shock cells at 42C for 45 sec and immediately place on ice
-
Mix 500 ul outgrowth media with cells and plate 100 ul
Resources
Lab notebook
We received the resuspended oligonucleotides from Noah, our TA for this assignment. First we diluted the oligo pairs in PCR compatible eppendorf tubes, 80 ul of water + 10 ul of each oligo, that way they would annealed and make duplexes.
Then we prepared the Golden gate annealed duplex fragments that would react as PCR primers mixing each oligo pair dilution (8ul) with T4 DNA ligase Buffer(1ul) and T4 Polynucleotide Kinase (1ul). We left the tubes in the thermocycler and programmed it for 37C incubation for 1 hour, 95C heating for 5 minutes and lastly gradually cool at Room Temperature (~25C) for another hour.
During this process the T4 PNK catalyzes the transfer and exchange of Pi from the γ position of ATP to the 5 ́ -hydroxyl terminus of polynucleotides. PNK also catalyzes the removal of 3 ́-phosphoryl groups from 3 ́-phosphoryl polynucleotides. T4 PNK requires ATP for activity, but its supplied reaction buffer does not contain ATP because it interferes with radiolabeling reactions. Typically, a kinase reaction is followed by a ligation reaction. To simplify this process, T4 PNK is optimized for use in T4 DNA Ligase reaction buffer (which contains the appropriate amount of ATP). That way we can then proceed to ligation without a buffer change or heat inactivation.
Then we prepared the other Golden Gate fragments by mixing the primer pairs (1,25 ul each) with a GFP template (1ul), dNTPs (1ul), Phusion Polymerase Buffer (10ul), Phusion DNA Polymerase (1ul), and water (34,5ul). We ran the PCR reaction following the program below in the thermocycler:
-
Initial denaturation - 1 cycle (98C for 30 seconds)
-
PCR - 30 cycles
1.Denaturation (98C for 10 seconds)
2.Annealing (60C for 30 seconds)
3.Extension (72C for 30 seconds)
-
Final extension - 1 cycle (72C for 5 minutes)
During a typical PCR, cycles of denaturation, annealing and extension are repeated to achieve exponential amplification of the target sequence. Denaturation consists of heating the samples up to a high temperature (typically 94-98°C) to cause denaturation of the template DNA, disrupting the hydrogen bonds and base stacking interactions that hold the DNA strands together. Once the strands are separated, the temperature is decreased to the annealing temperature to allow the primers to base pair (or anneal) to complimentary regions of the template. The annealing temperature is typically between 48-72°C. During the extension step (typically 68-72°C) the polymerase extends the primer to form a nascent DNA strand. This process is repeated multiple times (typically 25-35 cycles). The final step of the PCR is generally a longer, single temperature step (often 5-10 min at 68-72°C) that allows for the completion of any partial copies and the clearance of all replication machinery from the nascent DNA.
DNA Polymerase helps correct DNA sequences during amplification, lowering the error rate. Once the program finished, we ran the PCR reactions on agarose gel electrophoresis. We cut the gel and then extracted the correct-sized bands with a Gel Extraction Kit from Qiagen (centrifuge in tubes with columns 3 times, the first time with the extraction buffer and the other 2 times with water). The DNA extracted was measured in a Nanodrop in order to confirm the amplification during PCR.
After this process we combined and assembled the Golden Gate reactions mixing each fragment derived from pGreen_n(G/B/C)FP_Golgat_1F from the previous step with the primers (1ul each), Cutsmart Buffer (2ul), ATP (2ul), T7 DNA ligase (1ul), BbsI-HF (1ul) and water (8ul).
We ran these assemblies in the thermocycler following the program below:
1st Stage: 30 cycles
-
Digest (37C for 5 minutes)
-
Ligate (16C for 5 minutes)
2nd Stage: 1 cycle
-
Heat denature (55C for 10 minutes)
Once we finished this step, we proceeded to the bacterial transformation. We thawed 25ul of E. coli competent cells in ice for 30 minutes. We added 2ul of our Golden Gate Assembly to the cells and kept them on ice for 15 more minutes. Then we gave the cell a heat shock of 42C for 45 seconds in a water bath and immediately placed them on ice. We mixed 500ul of outgrowth media with the cells. And finally plated 100ul of each sample in Petri dishes filled with culture medium and antibiotics.

LAB TASK
EXPERIMENTAL HOMEWORK
Theory Task: Design a gene
Describe a detailed workflow for constructing and expressing it. Identify how the parts of your genetic construct relate to DNA replication and the Central Dogma of Molecular Biology.
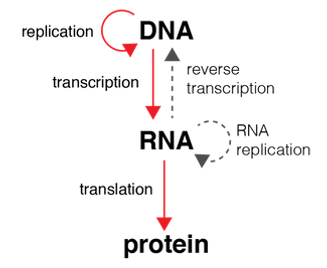
The Central Dogma of Molecular Biology is shown in the diagram on the right.
Genetic information flows into proteins by the following order: DNA → RNA → protein. Transcription is the process of copying a segment of DNA into mRNA. Translation is the process that synthesizes the language of nucleic acids to the language of proteins, or more precisely, amino acids that come together to make proteins. RNA is synthesized by the enzyme RNA polymerase.
The workflow for designing a gene is as follows:
-
Choose an application: what do you want the gene to do?
-
Find a scientific paper that describes the metabolic pathway
-
Choose the chassis: this is the host organism
-
Select the necessary parts from the iGEM Parts Registry
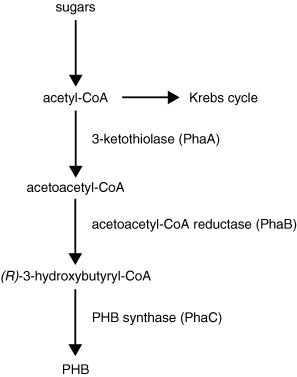
The chosen function of the gene is to produce a bio-derived and biodegradable plastic, namely the polymer polyhydroxybutyrate (PHB). PHB is a type of polyhydroxyalkanoate, or PHA [1]. PHAs are biodegradable thermoplastics that are synthesized by many different types of bacteria. Often, E.coli is chosen as the chassis but in this case PHB is produced by the bacteria Ralstonia eutrophus, Methylobacterium rhodesianum or Bacillus megaterium. The metabolic pathway to PHB is shown below.
The paper Bacterial synthesis of biodegradable polyhydroxyalkanoates [2] describes that this can be done with engineered E.Coli as well. "Genetic engineering is a powerful tool in the optimization of the microbial metabolism towards polymer production. Escherichia coli strains (Park et al. 2005a) have been genetically modified to produce PHB with an Mw up to 107 Da from glucose. This so-called ultra high molecular weight PHB (UHMW-PHB) can be processed into very strong films (Kahar et al. 2005)".
In the iGEM registry, a biological part (or simply, part) is a sequence of DNA that encodes for a biological function. As described in the Bio Design homework, we need a promoter, ribosome binding site (RBS), protein coding sequences (CDS) and a terminator.
The transcription unit is made by the promotor, protein coding sequence and terminator. A promoter is a sequence of DNA to which the RNA Polymerase binds when it starts transcription. The Open Reading Frame (ORF) is the coding sequence. After the ORF a terminator is added, which tells the RNA Polymerase to stop transcribing RNA, leave the DNA and prevents it from reading the next gene. The translation from the RNA unit into a protein is done by a ribosome binding site. This is the place where the ribosome attaches to the RNA message so it can start linking the specified amino acids into a protein chain.
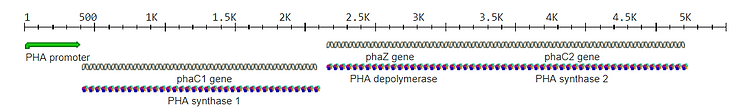
From the iGEM registry, the following parts were selected for constructing the gene:
-
Promoter: a constitutive promoter such as T7
-
Ribosome Binding Site: strong T7 RBS to match the promoter and terminator. Part:BBa_K1362090
-
Protein Coding sequences: Part:BBa_K1112002:Design
-
Terminator: nos, or a terminator from coliphage T7
Discussion
For someone who is new to the field of synthetic biology, it is challenging to navigate through the iGEM repository and design a gene from scratch. In the repository it is not indicated if a protein coding sequence is compatible with certain promoters, terminators and ribosome binding sites. When there is a lack of prior knowledge about these fundamentals, it is difficult to make the connection and holistically connect the correct parts to build up a gene. Therefore, it should be recognized that the information presented on this page is an incomplete and most likely incorrect representation of how to design a gene.
Protein Coding sequences: Part:BBa_K1112002:Design
Sources
-
R. Lenz, R. Marchessault. Biomacromolecules. 6, 1-8 (2005).
-
R.A.J. Verlinden, D.J. Hill, M.A. Kenward, C.D. Williams and I. Radecka (2006). Bacterial synthesis of biodegradable polyhydroxyalkanoates (Review Article) http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2672.2007.03335.x/pd
-
Kahar, P.,Agus, J., Kikkawa, Y., Taguchi, K., Doi, Y., Tsuge, T. (2005). Effective production and kinetic characterization of ultra-high-molecular-weight poly[(R)-3-hydroxybutyrate] in recombinant Escherichia coli.
-
Si Jae Park, Young-Ah Jang, HyukLee, A-Reum Park, Jung Eun Yang, Jihoon Shin, Young Hoon Oh, Bong Keun Song, Jonggeon Jegal, Seung Hwan Lee, Sang Yup Lee (2013). Metabolic engineering of Ralstonia eutropha for the biosynthesis of 2-hydroxyacid-containing polyhydroxyalkanoates
-
iGEM parts http://parts.igem.org/Part:BBa_K2260001
-
George Guo-Qiang Chen (2010). Plastics from Bacteria: Natural Functions and Applications. Springer International Publishing
In the paper of Kahar et al [3], recombinant Escherichia coli XL1-Blue was used as a chassis. Two expression plasmids harbouring Ralstonia eutropha P(3HB) biosynthesis genes (phaCAB(Re)) downstream of an inducible trc promoter, pTrcphaCAB(Re) and pJRDTrcphaCAB(Re) were constructed using the high copy number plasmid pTrc99a and the low copy number plasmid pJRDTrc1.
After entering the described plasmids and promoters in the iGEM registry, there were no results matching the query. The next step was to search for PHBs or PHAs in the iGEM registry. The composite part: BBa_K2260001 consists of phaC1 (Pseudomonas aeruginosa) and phaJ4 (Pseudomonas putida), which allow E. coli to convert short- and medium-chain-length volatile fatty acids (VFAs) to PHB. This part was inserted into pET29(b)+ downstream a T7 promoter and lacZ repressor. Because it is difficult to find what the exact elements are in this composite, I started searching for individual promoters, coding sequences, binding sites, and terminators.
Part BBa_K1112002:Design includes a protein coding sequence, that can be seen in the image below. The promoter is labeled as 'PHA promoter', which seems to be a generalization. In the papers, various types of promoters are used. The book Plastics from Bacteria: Natural Functions and Applications describes that the first report of PHB production in a high biomass crop was in a Saccharum sp. (sugarcane). Each gene phaA, phaB, and phaC from Ralstonia eutropha, was driven by the maize polyubiquitin promoter (ubi1) and nos terminator sequences and was cloned in a separate transformation vector. Other promoters listed in this book are cab-m5, Prrrn and psbA.
Recode green fluorescent protein (GFP) to instead emit blue or cerulean light. Confirm success by transforming your assembled BFP and CFP variants into E coli cells.
 |  |  |  |  |
|---|---|---|---|---|
 |  |  |  |  |
 |  |  |  |  |
 |  |